
Porous Materials
Energetics of Small Hydrocarbons in Metal Organic Frameworks
Selective sorption of hydrocarbons by tunable sorbents such as metal-organic frameworks (MOF) is among the most promising alternative to traditional cryogenic distillation. In our investigations, we apply density functional theory (DFT, a posteriori corrected to include the description of long range dispersion forces) to investigate the selective sorption of C2-C4 hydrocarbons in MOF-74-Mg/Zn.
We use molecular clusters and periodic boundary conditions (PBC) to assess the reliability of the models and explain possible fortuitous result agreements. While both approaches agree in showing significant differences in binding energies between olefins and paraffins at the open metal sites of the MOF, the binding energies found using molecular cluster models are significantly smaller than those obtained from the periodic approach. This aspect seems to indicate the importance of fully accounting for the chemical environment experienced by the adsorbed hydrocarbons.
In fact, when PBC models are used to investigate the interaction of the small hydrocarbons with the MOF open metal sites, not only the overall chemical environment of the frameworks is included, but this allows correcting the binding energies for basis-set superposition error, molecular lateral interaction, zero-point energy, and thermal contributions. As such, our approach is unique as it allows for a direct comparison with respect to experimental calorimetric values.
In our works (see references below) we isolated for the first time the origin of the fictitious agreement between binding energies obtained with molecular cluster models and experimental heats of adsorption, identifying its origin as due to compensation of errors.
In addition, our differential enthalpies of adsorption very well agree with available experimental data, further strengthening the reliability of our approach.
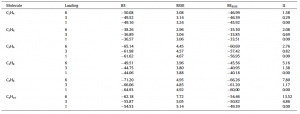
Binding energy (BE), basis set superposition error (BSSE) correction to the BE, BSSE corrected BE (BEBSSE), and lateral interaction (LI) computed for adsorbed molecules in MOF-74-Mg with respect to different loadings. All the energies are reported in kJ/mol.
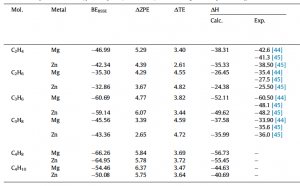
Binding energies (BEBSSE), zero-point energy (DZPE), and thermal energy correction (DTE) providing heats of adsorption (DH) of C2-C4 for high molecular loadings in both MOF-74-Mg/Zn. Available experimental results are also reported [44,45] to assess the differential enthalpy for the same molecule between the two MOFs, and for the same MOF with respect to the olefin/paraffin pairs. All the energies are reported in kJ/mol.
References
D. Degaga (grad student), L. Valenzano*, Part II: Quantum mechanical prediction of heats of adsorption for C2-C4 hydrocarbons in MOF-74-Mg/Zn periodic structures, 2017, Chemical Physics Letters, 682, 168-174
D. Degaga (grad student), L. Valenzano*, Part I: C2-C4 Hydrocarbons Separation Addressed via Molecular Cluster Models Carved Out From Periodic MOF-74-Mg/Zn Structures, 2016, Chemical Physics Letters, 660, 313-319
Diffusion Mechanisms of Small Hydrocarbons in Metal Organic Frameworks
In this project we adopt the climbing-image nudge elastic band (CI-NEB) method coupled with density functional theory to model the molecular diffusion of CH4 (methane), C2H2 (acetylene), C2H4 (ethylene), C2H6 (ethane), and H2O (water) in MOF-74-Mg.
At the moment we are concentrating on the description of three different diffusion mechanisms: two within the MOF cavity, and one along the channels of the open metal sites (see figures below).
Together with our recent investigations on heats of adsorption, lateral interaction, and IR frequency shifts, this work provides an important addition toward a comprehensive description of adsorption and diffusion processes of small hydrocarbons in MOF-74-Mg.
Spectroscopic and Thermodynamic Properties of Hydrocarbons in Metal Organic Frameworks